指导原则
整形用面部植入假体注册技术 审查指导原则(2020年第36号通告附件4)
本指导原则旨在为药品监管部门对注册申报资料的技术审评提供技术指导,同时也为注册申请人进行整形用面部植入假体的产品注册申报提供参考。
本指导原则系对整形用面部植入假体的一般要求,注册申请人应依据具体产品的特性对注册申报资料的内容进行充实和细化,并依据具体产品的特性确定其中的具体内容是否适用。
本指导原则是对注册申请人和审评人员的技术指导性文件,不包括注册审批所涉及的行政事项,亦不作为法规强制执行。如果有其他科学合理的替代方法,也可以采用,但是需要提供依据及相关资料。应在遵循相关法规和标准的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制订的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
一、适用范围
本指导原则所涉及的整形用面部植入假体是指人工合成不可吸收材料制备的,用于面部填充的,仅以物理占位作用机理获得整形效果的产品。具体产品的适用范围需根据产品性能特点及临床数据进行确定。目前境内已上市的该类产品材质主要有硅橡胶、膨体聚四氟乙烯/全氟乙丙烯、高密度聚乙烯等。
其他材料制备的整形用植入假体可参考本指导原则中适用的部分。
二、技术审查要求
注册申报资料按照国家药品监督管理局医疗器械注册相关规章进行提供,尤其注意以下几方面内容:
(一)产品名称
根据《医疗器械分类规则》(国家食品药品监督管理总局令第15号)、《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)等相关文件确认产品名称并论述其确定依据。该类产品通用名一般为“整形用面部植入假体、面部整形植入填充材料”。可添加材料化学名称,如“膨体聚四氟乙烯面部植入整形填充材料”;可反映具体植入部位,如“植入性硅橡胶鼻假体”。
(二)注册单元划分
根据《医疗器械注册单元划分指导原则》(国家食品药品监督管理总局通告2017年第187号)对申报产品的注册单元进行确认,原则上材料成分不同的整形用面部植入假体应划分为不同的注册单元,如硅橡胶与膨体聚四氟乙烯产品作为不同的注册单元;硅橡胶成分配比、硫化程度不同时作为不同的注册单元。
(三)产品综述及基本信息
1.详述产品作用原理,预期与人体接触部位(解剖部位)、接触方式、作用时间。
2.明确列出终产品中所有材料成分及其含量,包括规范的化学名称、化学结构式/分子式、材料商品名(若有)、材料代号(若有)等。
3.详述产品总体外形、尺寸描述及结构(提供相应图示):形状、尺寸范围及间隔(包括长度、宽度、高度、其他特征尺寸)等。明确产品型号、规格间的异同点(同一型号的产品需具有材料、性质、结构上的同一性)。对于根据临床需要而对表面进行特殊加工或处理的植入体,需给出表面形态的信息(提供相应图示)。
建议按照下表格式列出产品的基本信息(可根据具体情况增减项目):
|
型号、规格 |
表面形态 |
长度(mm) |
宽度(mm) |
高度(mm) |
其他特征尺寸 |
|
XXXX |
光滑/多孔 |
|
|
|
|
图示举例:
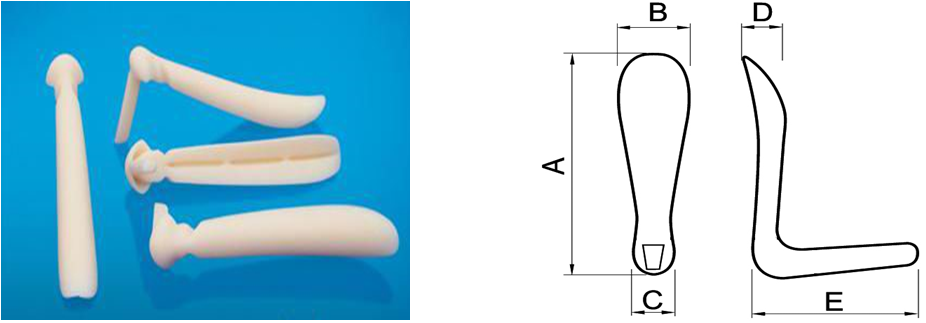
5.产品适用范围和禁忌症:注册申请人应根据临床评价资料规范申报产品的适用范围和禁忌症描述。如适用范围描述为该产品适用于鼻背、颏部及颞部凹陷的填充。
6.提供产品的境内外动态分析情况(包括境内外同类产品的上市情况及与申报产品工作原理、结构组成、制造材料、性能指标、适用范围等情况的对比)。
(四)产品原材料相关审评要求
1.详述产品所用原材料(包括整个生产过程中加入的溶剂、催化剂、交联剂、抗氧化剂、脱模剂、稀释剂、色素等添加剂/助剂等)的规范的材料化学名称、化学结构式/分子式、材料商品名(若有)、材料代号(若有)及有关安全使用的支持性资料。
2.若原材料外购,需明确原材料供应商并附其资质证明文件、供销关系证明文件(供销协议)、质量标准及测试报告。若原材料为自行合成,应提供材料生产过程中的质量控制标准及相关的验证报告。
对于适用相关材料国家标准/行业标准的原材料,应提供原材料符合标准的验证报告。如硅橡胶原材料,明确所采用的硫化形式(如双组分加成硫化、热硫化等)。对于采用双组分加成硫化的硅橡胶面部植入假体产品,应提供原材料符合YY0484《外科植入物 双组分加成型硫化硅橡胶》的验证资料。
(五)产品适用的相关标准
主要参考标准举例(未标明年代号表示应参照最新标准):
GB/T 16886《医疗器械生物学评价》系列标准
GB/T 16175《医用有机硅材料生物学评价试验方法》
GB/T 528《硫化橡胶或热塑性橡胶拉伸应力应变性能的测定》
GB/T 529《硫化橡胶或热塑性橡胶撕裂强度的测定(裤型、直角形和新月形试样)》
GB/T 531.1《硫化橡胶或热塑性橡胶 压入硬度试验方法 第1部分:邵氏硬度计法(邵尔硬度)》
GB/T 531.2《硫化橡胶或热塑性橡胶 压入硬度试验方法第2部分:便携式橡胶国际硬度计法》
GB/T 14233.1《医用输液、输血、注射器具检验方法 第1部分:化学分析方法》
GB/T 14233.2《医用输液、输血、注射器具检验方法 第2部分:生物学试验方法》
YY/T 0640《外科植入物 通用要求》
YY 0334《硅橡胶外科植入物通用要求》
YY0484《外科植入物双组分加成型硫化硅橡胶》
《中华人民共和国药典》
ASTM F754-08 Standard Specification for Implantable Polytetrafluoroethylene (PTFE) Sheet, Tube, and Rod Shapes Fabricated from Granular Molding Powders
(六)产品研究资料
1.产品性能研究
(1)详述产品技术要求中性能指标及检验方法的确定依据,提供采用的原因及理论基础,提供涉及到的研究性资料、文献资料和/或标准文本。
(2)提供不同批次产品的力学性能的研究资料,样本量需有统计学考虑。研究资料应详细明确各项目指标可接受的标准、具体试验方法、验证样品批次及样本量、试验结果数据及试验结论等。详述性能研究资料中性能指标及检验方法的确定依据,
(3)对于硅橡胶材质的假体,提供三个不同硫化批次硅橡胶硫化程度的测试报告,以考查不同批次样品的硫化程度的均匀一致性。测定硫化程度的方法有:测量低应变下的杨氏模量(与硫化程度成正比),或在良溶剂中测量聚合成分的平衡溶胀率,或测定总浸提物中未反应的硫化剂等。
(4)对于硅橡胶材质的假体,提供可沥滤物如D4、D5等小分子物质限量控制的研究资料,需采用极限浸提的方法。
(5)测试最终产品中任何有潜在毒性、致癌性的材料成分含量,如有机及无机杂质等,并提供以上物质的人体接触许可限量/阈值及其确定依据。
(6)应提供上述研究资料中试验样品所用型号、规格的典型性分析资料。
2.生物相容性评价研究
需对终产品中与患者和使用者直接或间接接触的材料的生物相容性进行评价。
生物相容性评价研究资料需包括:
(1)生物相容性评价的依据和方法。
(2)产品所用材料的描述及与人体接触的性质。
(3)实施或豁免生物学试验的理由和论证。
(4)对于现有数据或试验结果的评价。
整形用面部植入假体需考虑的生物相容性评价项目包括:细胞毒性、致敏、刺激或皮内反应、急性全身毒性、亚慢性毒性、遗传毒性、植入后局部反应等。建议按照有效的GB/T 16886标准和其他生物学评价相关文件进行生物学评价。
若申报产品中的材料从未在境内已上市的长期植入性医疗器械中使用,需提供该材料适合用于人体使用的相关支持性资料并应对材料的长期生物相容性进行评价,如远期植入后反应、慢性毒性、致癌性、基于可沥滤物分析基础上的毒代动力学研究等。
3.灭菌工艺研究
产品需经最终灭菌,明确灭菌工艺(方法和参数)和无菌保证水平(SAL),SAL需达到10-6,提供灭菌确认报告。如灭菌使用的方法容易出现残留,需明确残留物信息及采取的处理方法,并提供研究资料。
4.产品有效期和包装研究
提供产品有效期的验证报告(包括产品物理、化学稳定性和包装密封稳定性的验证资料)。不同包装或容器的产品需分别提供验证资料。
(七)生产制造信息
1.详述产品生产加工过程,明确产品生产加工工艺,注明关键工艺和/或特殊过程,并说明其过程控制点。明确产品生产过程中各种加工助剂的使用情况及对各种有机、无机杂质(如残留单体、小分子残留物、重金属等)的控制情况并提交相应的验证资料。提供涉及产品安全性的加工工艺的确定依据以及涉及到的研究性资料、文献资料等。
2.有多个研制、生产场地的,需概述每个研制、生产场地的实际情况。
(八)产品的风险分析资料
根据YY/T 0316《医疗器械 风险管理对医疗器械的应用》,充分识别面部植入假体产品的原材料、生产加工过程、产品包装、灭菌、运输、贮存、使用等产品生命周期内的各个环节的安全特征,从生物学危险(源)、环境危险(源)、有关临床使用的危险(源)、由功能失效及老化引起的危险(源)等方面,进行全面的风险分析及风险评价,并详述所采取的风险控制措施。在所有风险控制措施已经实施并验证后,注册申请人应对综合剩余风险是否可接受进行评价并给出结论性意见。
(九)临床评价资料
按照《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号)提交临床评价资料。关于该类产品临床评价(包括临床试验)的具体要求请参考临床评价相关规章及指南文件。
(十)产品技术要求
1.产品相关信息
(1)产品基本信息及图示的要求可参考综述资料。
(2)明确产品各组成部分的所有组成材料的基本信息,如:规范的化学名称、化学结构式/分子式、分子量及其分布(如硅橡胶则提供硫化前的数据)、材料代号/商品名/牌号(如适用)等。明确原材料硅橡胶所采用的硫化形式(如双组分加成硫化、热硫化等)及硫化程度(如适用)。
2.项目要求
(1)外观及尺寸:长度、宽度、高度、其它特征尺寸。
(2)表面特性:对于根据临床需要而对表面进行特殊加工或处理的植入体,需给出表面形态的要求及测试方法。
(3)根据产品的适用范围、材料组成及设计特点制定适用的物理性能要求:如孔隙率、拉伸强度、扯断伸长率、撕裂强度、粘接强度(对于多个组件粘接的产品)、邵尔硬度(硅橡胶材质)等。
(4)根据产品的材料组成及设计特点制定适用的化学性能要求:如蒸发残渣、酸碱度、还原物质、紫外吸光度、重金属总量、终产物中有毒小分子物质残留量要求等。
对于硅橡胶材料的产品,还需考虑干燥失重、微量元素(铅Pb、镉Cd、砷As、铬Cr、铁Fe)、过氧化物(热硫化硅橡胶材质)、极限浸提小分子物质(如D4、D5)。
对于聚四氟乙烯材料的产品,还需考虑溶剂残留量(如D80)、四氯化碳浸提试验(浸提后样品外观、可浸提碳氢化合物红外分析)、蒸馏水浸提试验(浸提后样品外观、水浸提液的电阻率)。
对于经环氧乙烷(EO)灭菌的产品,应制定EO残留量要求。
(5)无菌。
3.上述若有不适用的项目需在研究资料中详细说明理由。对于无法在终产品中测定的项目需提供充分理由并在研究资料中提供中间品相关性能的质控资料。除上述要求外,还需参照相关材料的国家标准/行业标准、药典以及根据产品自身技术特点增加适用的化学性能要求。
(十一)产品说明书及标签
产品说明书及标签样稿应符合《医疗器械说明书和标签管理规定》(国家食品药品监督管理总局令第6号)、YY/T 0640《无源外科植入物通用要求》等相关法规及标准的要求,此外需注意:
1.需注明“该产品仅限于在国家正式批准的医疗机构中由具有相关整形专业医师资格的人员,严格按照产品使用说明书的要求进行使用”。
2.性能特征描述应以企业提交的注册资料为准。
3.产品适用范围需与临床验证过的范围一致,另外需明确填充的具体解剖部位。
4.对于临床评价资料中涉及的禁忌症或注意事项需在说明书中给予提示。
5.产品货架有效期、保存运输条件需与技术支持性资料一致。
6.说明书中不应含有夸大宣传的相关描述、未经验证及支持的有关内容。
三、参考资料
1. GB/T 16886《医疗器械生物学评价》系列标准
2. GB/T 16175-2008《医用有机硅材料生物学评价试验方法》
3. GB/T 528-2009《硫化橡胶或热塑性橡胶 拉伸应力应变性能的测定》
4. GB/T 529-2008《硫化橡胶或热塑性橡胶撕裂强度的测定(裤形、直角形和新月形试样)》
5. GB/T 531.1-2008《硫化橡胶或热塑性橡胶 压入硬度试验方法 第1部分:邵氏硬度计法(邵尔硬度)》
6. GB/T 531.2-2009《硫化橡胶或热塑性橡胶 压入硬度试验方法第2部分:便携式橡胶国际硬度计法》
7. GB/T 14233.1-2008《医用输液、输血、注射器具检验方法 第1部分:化学分析方法》
8. GB/T 14233.2-2005《医用输液、输血、注射器具检验方法 第2部分:生物学试验方法》
9. YY/T 0640-2016《无源外科植入物 通用要求》
10. YY 0334-2002《硅橡胶外科植入物通用要求》
11. YY0484-2004《外科植入物 双组分加成型硫化硅橡胶》
12.《中华人民共和国药典》2015版
13. ASTM F754-08(2015) Standard Specification for Implantable Polytetrafluoroethylene (PTFE) Sheet, Tube, and Rod Shapes Fabricated from Granular Molding Powders
14.《无源植入性医疗器械产品注册申报资料指导原则》(食药监办械函〔2009〕519号)
15.《无源植入性医疗器械货架有效期注册申报资料指导原则(2017年修订版)》(国家食品药品监督管理总局通告2017年第75号)
16.《医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则》(国家药品监督管理局通告2019年第78号)
17.《医疗器械动物实验研究技术审查指导原则 第一部分:决策原则》(国家药品监督管理局通告2019年第18号)
18.《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号)
19.《接受医疗器械境外临床试验数据技术指导原则》(国家食品药品监督管理总局通告2018年第13号)
四、编写单位
本指导原则由国家药品监督管理局医疗器械技术审评中心编写并负责解释。
录入:2021/2/20 16:50:22 点击:1638

